уравнения состояния
УРАВНЕНИЯ СОСТОЯНИЯ
уравнения, выражающие связь между параметрами состояния физически однородной системы при термодинамич. равновесии. Термическое У.с. связывает давление р с объемом V и температурой T, а для многокомпонентных систем — также с составом (молярными долями компонентов). Калорическое У. с. вьюажает внутр. энергию системы как функцию V, T и состава. Обычно под У. с., если специально не оговаривается, подразумевают термич. У. с. Из него можно непосредственно получить коэф. термич. расширения, коэф. изотермич. сжатия, термич. коэф. давления (упругости). У. с. является необходимым дополнением к термодинамич. законам. Пользуясь У. с., можно раскрыть зависимость термодинамич. функций от V и р, проинтегрировать дифференц. термодинамич. соотношения, рассчитать летучести (фугитивности) компонентов системы, через которые обычно записывают условия фазового равновесия. Термодинамика устанавливает связь между У. с. и любым из термодинамических потенциалов системы, выраженным в виде функции своих естественных переменных. Например, если известна энергия Гельмгольца 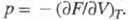 (свободная энергия) F как функция T и V, то У. с. не может быть получено с помощью одних только законов термодинамики, оно определяется из опыта или выводится методами статистич. физики. Последняя задача очень сложная и м. б. решена лишь для упрощенных моделей системы, напр., для идеального газа. У. с., применяемые для реальных систем, имеют эмпирич. или полуэмпирич. характер. Ниже рассмотрены некоторые наиб. известные и перспективные У. с.
(свободная энергия) F как функция T и V, то У. с. не может быть получено с помощью одних только законов термодинамики, оно определяется из опыта или выводится методами статистич. физики. Последняя задача очень сложная и м. б. решена лишь для упрощенных моделей системы, напр., для идеального газа. У. с., применяемые для реальных систем, имеют эмпирич. или полуэмпирич. характер. Ниже рассмотрены некоторые наиб. известные и перспективные У. с.
У. с. идеального газа имеет вид pV=RT, где V — молярный объем, R — универсальная газовая постоянная. Этому уравнению подчиняются реальные газы при высоких разрежениях (см. Клапейрона — Менделеева уравнение).
Свойства реальных газов при небольших и средних давлениях хорошо описываются вириальным уравнением: pV/RT = 1 + B2 /V+B3 /V2 + ..., где B2, В3 — второй, третий и т. д. вириальные коэффициенты. Для данного вещества они зависят лишь от температуры. Вириальное У. с. обосновано теоретически; показано, что коэф. B2 определяется взаимод. пар молекул, В3 — взаимод. трех частиц и т. д. При больших плотностях вещества записанное выше разложение по степеням обратного объема расходится, поэтому вириальное уравнение непригодно для описания жидкостей. Оно служит лишь для расчета летучестей компонентов газообразных веществ. Обычно ограничиваются членом B2 /V (редко B3/V2). В лит. приводят эксперим. значения вириальных коэф., разработаны и теоре-тич. методы их определения. У. с. со вторым вириальным коэф. B2 широко используют для моделирования газовой фазы при расчетах фазовых равновесий в случае не слишком высоких давлений (до 10 атм). Его применяют также для описания свойств разбавленных растворов высокомол. веществ (см. растворы полимеров).
Для практич. расчетов фазовых равновесий в широком диапазоне температур и давлений важное значение имеют У. с., способные описать одновременно свойства жидкой и газовой фаз. В первые такое уравнение было предложено И. Ван-дер-Ваальсом в 1873:
р = RT(V-b)-a/V2,
где а и b — постоянные Ван-дер-Ваальса, характерные для данного вещества (см. Ван-дер-Ваальса уравнение). Это У. с. имеет третий порядок относительно объёма V, любая изотерма при параметрах состояния, меньших критич. значений (в докри-тич. области), имеет три действит. положит, корня при фиксир. давлении. Наиб. из корней уравнения соответствует газовой фазе, наименьший — жидкой; средний корень уравнения физ. смысла не имеет. В сверхкритич. области параметров состояния изотермы имеют лишь один действит. корень.
Кубич. зависимость давления от объема сохраняется во MH. эмпирич. модификациях уравнения Ван-дер-Ваальса. Чаще других используют двухпараметрич. уравнения Пенга — Робинсона (1976) и Редлиха — Квонга — Соаве (1949, 1972). Эмпирич. постоянные этих У. с. можно определить по критич. параметрам вещества (см. критическое состояние). Чтобы расширить круг описываемых У. с. систем, набор рассматриваемых свойств, диапазон температур и давлений, разработаны кубич. У. с., содержащие три и более эмпирич. постоянных. Важное преимущество кубич. У. с. — их простота, благодаря чему при расчетах с помощью ЭВМ не требуется слишком больших затрат машинного времени. Для мн. систем, образованных неполярными или слабо полярными веществами, эти У. с. обеспечивают требуемую для практич. целей точность.
Если известны подробные эксперим. данные о р-V-T-зависимостях, для их обобщения привлекают многопараметрич. эмпирические У. с. Одно из наиб. распространенных У. с. такого типа — уравнение Бенедикта — Веббa Pубина (уравнение БВР), разработанное в 1940 на основе вириального У. с. В этом уравнении давление р представлено в виде полинома плотности вещества с коэффициентами, зависящими от температуры. Членами ряда высоких порядков пренебрегают, а для компенсации включают в уравнение экспоненциальный член. Это приводит к появлению S-образных изотерм и дает возможность описывать жидкую фазу и равновесия жидкость — газ.
Для неполярных и слабо полярных веществ уравнение БВР дает очень точные результаты. Для индивидуального вещества оно содержит восемь подгоночных параметров, для смеси дополнительно вводятся параметры смешанного ("бинарного") взаимодействия. Оценка большого числа подгоночных параметров — задача очень сложная, требующая многочисленных и разнообразных эксперим. данных. Параметры уравнения БВР известны лишь для неск. десятков веществ, гл. обр. углеводородов и неорг. газов. Модификации уравнения, направленные, в частности, на повышение точности описания свойств конкретных веществ, содержат еще большее число подгоночных параметров. Несмотря на это, добиться удовлетворит, результатов для полярных веществ не всегда удается. Усложненность формы затрудняет использование У. с. этого типа при расчетах процессов дистилляции, когда необходимо выполнять многократную оценку летучестей компонентов, объема и энтальпии системы.
При описании смесей веществ эмпирич. постоянные У. с. считаются зависящими от состава. Для кубич. У. с. ван-дер-ва-альсового типа общеприняты квадратичные правила смешения, согласно которым постоянные а и b для смеси определяют из соотношений:
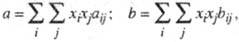
где xi, xj — молярные доли компонентов, величины aij и bij связывают с постоянными для индивидуальных веществ aii, ajj и bii, bjj согласно комбинационным правилам:
aij = (aiiajj)1/2(1-kij); 6ij = (bii+bjj)/2,
где kij — подгоночные параметры смешанного взаимод., определяемые по эксперим. данным. Однако квадратичные правила смешения не позволяют получить удовлетворит, результаты для т. наз. асимметричных систем, компоненты которых сильно отличаются по полярности и мол. размерам, напр., для смесей углеводородов с водой.
M. Гурон и Дж. Видал в 1979 сформулировали правила смешения нового типа, опирающиеся на модели локального состава, которые успешно передают асимметрию концентрац. зависимостей избыточного потенциала Гиббса GE для жидких смесей и позволяют существенно улучшить описание фазовых равновесий. Суть подхода состоит в том, что приравнивают величины GE жидкого раствора, получаемые из У. с. и рассчитываемые согласно выбранной модели локального состава [уравнения Вильсона, NRTL (Non-Random Two Liquids equation), UNIQAC (UNIversal QUAsi-Chemical equation), UNIFAC (UNIque Functional group Activity Coefficients model); см. растворы неэлектролитов]. Это направление интенсивно развивается.
Многие двухпараметрич. У. с. (Ван-дер-Ваальса, вириальное с третьим вириальным коэф. и др.) можно представить в виде приведенного У. с.:
f(pпр, Тпр, Vпр)= 0,
где pпр = р/ркрит, Тпр=Т/Ткрит, Vпр= V/Vкрит — приведенные параметры состояния. Вещества с одинаковыми значениями рпр и Тпр имеют одинаковый приведенный объем Vnp; совпадают также факторы сжимаемости Z = pV/RT, коэф. летучести и некоторые др. термодинамич. функции (см. соответственных состояний закон). Более общий подход, который позволяет расширить круг рассматриваемых веществ, связан с введением в приведенное У. с. дополнит, параметров. Наиб. простые среди них — фактор критич. сжимаемости Zкрит = ркритVкрит/RTкрит. и ацентрич. фактор w= -Ig pпр −1 (при Тпр = 0,7). Ацентрич. фактор является показателем несферичности поля межмол. сил данного вещества (для благородных газов он близок к нулю).
К. Питцер предложил пользоваться для расчета фактора сжимаемости линейным разложением
Z(Tкрит, pкрит) = Z0(Tкрит, pкрит)+ wZ'(Tкрит, pкрит),
где Z0 означает фактор сжимаемости "простой" жидкости, напр., аргона, a Z' характеризует отклонения от модели простой жидкости (см. жидкость). Предложены корреляционные соотношения, определяющие зависимости Z°(Tкрит, pкрит)
и Z'(Tкрит, pкрит). Наиб. известны корреляции Ли и Кесслера, в которых зависимость Z0 от Tкрит и pкрит передается с помощью уравнения БВР для аргона. Зависимость Z' от Tкрит и pкрит установлена при выборе в качестве "эталонной" жидкости н-октана. Принимается, что Z'(Tкрит, pкрит) = [Z*(Tкрит, pкрит) — Z°(Tкрит, pкрит)]/w*, где w* — фактор ацентричности н-октана, Z* — его фактор сжимаемости согласно уравнению БВР. Разработана методика применения уравнения Ли — Кесслера и для жидких смесей. Это У. с. наиб. точно описывает термодинамич. свойства и фазовые равновесия для неполярных веществ и смесей.
Наряду с вышеупомянутыми эмпирич. У. с. важное значение приобрели уравнения, обладающие возможностями учета особенностей структуры молекул и межмол. взаимод. Они опираются на положения статистич. теории и результаты численных экспериментов для модельных систем. Согласно мол.-статистич. трактовке, уравнение Ван-дер-Ваальса описывает флюид твердых притягивающихся сфер, рассматриваемый в приближении среднею поля. В новых уравнениях уточняется прежде всего член уравнения Ван-дер-Ваальса, обусловливаемый силами межчастичного отталкивания. Значительно точнее приближение Кариахана — Старлинга, опирающееся на результаты численного моделирования флюида твердых сфер в широком диапазоне плотностей. Оно используется во многих У. с., однако большие возможности имеют У. с. модельных систем твердых частиц, в которых учитывается асимметрия мол. формы. Например, в уравнении BACK (Boublik-Alder-Chen-Kre-glewski) для оценки вклада сил отталкивания служит У. с. флюида твердых частиц, имеющих форму гантелей. Для учета вклада сил притяжения употребляют выражение, аппроксимирующее результаты, полученные методом мол. динамики для флюида с межчастичными потенциалами типа прямоугольной ямы (см. молекулярная динамика). Уравнение BACK и его аналоги позволяют с достаточной точностью описывать смеси, не содержащие высококипящих компонентов.
Особенность описания смесей высококипящих орг. веществ — необходимость учета дополнительной вращательно-колебат. степени свободы, связанной со смещениями сегментов молекул-цепочек (напр., алкенов C8). Для этих систем наиб. распространение получило уравнение PHCT (Perturbed Hard Chain Theory), предложенное Дж. Прауснитцем и М. Донахью в 1978. Индивидуальное вещество характеризуется тремя эмпирич. параметрами в уравнении PHCT. Комбинационные правила для смеси содержат один параметр смешанного взаимодействия. Дальнейшее усовершенствование уравнения PHCT основано на замене потенциала прямоугольной ямы, описывающей притяжение молекул, потенциалом Леннард — Джонса [уравнение PSCT (Perturbed Soft Chain Theory)] и на учете анизотропии межмол. сил [уравнение PACT (Perturbed Anisotropic Chain Theory)]. Последнее уравнение хорошо описывает фазовые равновесия в системах с полярными компонентами даже без использования подгоночных параметров парного взаимодействия.
Предложен ряд У. с., в которых в явном виде учитываются взаимод. молекул, приводящих, напр., к образованию водородных связей (ассоциация молекул), путем нахождения равновесных концентраций ассоциатов с помощью действующих масс закона.
В 80-х гг. появились т. наз. групповые У. с. [UNIWAALS (UNIfac van-der-WAALS equation), уравнение Скьолда — Йорген-сена, MHV-2, дырочное уравнение, разработанное H. А. Смирновой и А. И. Викторовым, и др.]. Они позволяют прогнозировать свойства широкого круга систем, зная модельные параметры для сравнительно небольшого числа структурных фрагментов (групп), из которых состоят молекулы компонентов.
Все возрастающий интерес к У. с. обусловлен прежде всего практич. потребностями разработки мн. совр. технологий, связанных с абсорбционным разделением веществ, эксплуатацией нефтяных и газовых месторождений и т. п., поскольку в этих случаях требуется количеств, описание и прогнозирование фазовых равновесий в широком диапазоне температур и давлений. Однако пока не существует достаточно универс. У. с. Все упомянутые У. с. оказываются неточными при описании состояний вблизи критич. точки и не предназначены для рассмотрения критических явлений. Для этих целей разрабатываются специальные У. с., но и они пока плохо приспособлены для конкретных практич. приложений.
У. с. твердых тел определяют, напр., зависимость модулей упругости от температуры и давления. Теоретич. расчеты свободной энергии и модулей упругости проводятся для сравнительно простых моделей твердого тела.
У. с. для систем, находящихся во внеш. поле, разрабатываются и исследуются в соответствующих разделах физики. В физике высоких давлений и температур на основе эксперим. данных и общих теоретич. представлений разработаны модели У. с., охватывающих все агрегатные состояния, включая плотную плазму.
Лит.: Рид Р., Прауснитц Дж., Шервуд Т., Свойства газов и жидкостей, пер. с англ., Л., 1982; У эйлес С., Фаловые равновесия в химической технологии, пер. с англ., ч. 1, М., 1989; Викторов А. И. (и д р.), "Ж. прикл. химии", 1991, т. 64, № 5, с. 961–78.
Г. Л. Куранов
