кислоты и основания
КИСЛОТЫ И ОСНОВАНИЯ
Термины "кислоты" и "основания" вполне сформировались в 17 в. Их содержание неоднократно пересматривалось и дополнялось. Этот процесс происходил и происходит в острых столкновениях представителей разных взглядов на природу К. и о. Развитие взглядов на К. и о. А. Лавуазье (1778) объяснял свойства кислот наличием в них кислорода ("кислородная теория" кислот). Однако скоро выяснилось, что очень мн. кислородсодержащие вещества (оксиды металлов, соли и др.) не обладают кислотными свойствами, а ряд типичных кислот, напр. соляная, не содержат кислорода (Г. Дэви и Ж. Гей-Люссак 1810, 1814). И. Берцелиус (1802–19) устранил первое из этих противоречий, приписав оксидам знак электрич. заряда. Электроотрицат. (по Берцелиусу) оксиды неметаллов образуют кислоты, электроположит. оксиды металлов — основания. В 1814 Дэви высказал мнение, что атом водорода — необходимая составная часть кислот. Ю. Либих (1833) уточнил эту "водородную теорию" кислот, показав, что кислотные свойства обусловлены не любым атомом водорода, а лишь тем, который способен замещаться металлом. После появления теории электролитич. диссоциации С. Аррениуса (1887) сформировалась ионная теория К. и о. Согласно этой теории, кислота — водородсодержащее соед., при электролитич. диссоциации которого в воде образуются ионы водорода и анионы, а основание-соед., диссоциирующее с отщеплением ионов гидроксила и катионов. В дальнейшем появились разл. варианты обобщения ионной теории К. и о. применительно к неводным растворителям. Эти варианты не противоречат, а дополняют друг друга, большинство их используется и разрабатывается в настоящее время. Э. Франклином в 1924 создана сольвентная теория. По этой теории, К. и о.-вещества, при растворении которых увеличивается концентрация соотв. катионов и анионов, образующихся при диссоциации растворителя. В этом случае кислотно-основное взаимод. выражается схемой: кислота + основание : соль + растворитель Сольвентная теория способствовала исследованию К. и о. в неводных растворах. Учитывая комплексообразование, А. Вернер (1907) предложил теорию ангидро- и аквакислот и оснований. Согласно этой теории, в водном растворе безводные кислоты, т. наз. ангидро-кислоты (А) и ангидрооснования (В), превращаются в аква-кислоты [АОН]−H+ и акваоснования [ВН]+OH−, которые диссоциируют:
[AOH]−H+:[АОН]−+H+
[ВН]+OH−:[ВН]++OH−
Хотя схемы, передающие механизм взаимод. с водой во мн. случаях неверны, взгляды Вернера дали некоторый импульс для изучения роли воды в кислотно-основном взаимод. Из работ в этом направлении выделяются работы А. Ганча (1917–27), создавшего т. наз. хим. теорию кислот. В этой теории кислоты определены как соед. водорода, в которых последний м. б. замещен на металл или неметаллоподобный радикал. Важнейший признак кислот — способность давать соли. Ионизация кислот в растворе происходит в результате их взаимод. с растворителем. Теория содержит принципиально новое положение: в растворах кислотные свойства проявляются не самой кислотой, а сольватир. катионами водорода. В хим. теории кислот четко сформулировано понятие об амфотерности — способности некоторых соед. проявлять как кислотные, так и основные свойства в зависимости от условий и природы реагентов, участвующих в кислотно-основном взаимодействии. В 1923 были предложены две, доминирующие по сей день, теории К. и о.: протонная теория И. Брёнстеда и Т. Лоури и электронная теория Г. Льюиса. По Брёнстеду , кислота — донор протона, а основание — акцептор его. По Льюису, кислота — вещество, которое может использовать неподеленную .пару электронов атома др. молекулы для образования устойчивой электронной группировки одного из своих атомов, основание — вещество, обладающее неподеленной парой электронов, которая м. б. использована для образования устойчивой электронной группировки др. атома. Часто такой группировкой является октет электронов. Теория, предложенная М.И. Усановичем (1939–53), объединяет электронную и протонную теории. По этой теории, кислота-вещество, способное отдавать катионы или присоединять анионы; основание — вещество, способное отщеплять анионы или присоединять катионы, напр.:
Fe(CN)3 (кислота) + 3KCN (основание):K3[Fe(CN)6] CH3I(кислота)+N(CH3)3 (основание):(CH3)4NI
Для всех обсуждаемых теорий характерно, что в них определения К. и о. зависят от определения понятия кислотно-основного процесса, в котором реагирующие между собой К. и о. являются таковыми лишь по отношению друг к другу. Единой теории кислотно-основного взаимод. и, следовательно, понятий К. и о. пока нет. В настоящее время наиб. широко используются две теории К. и о.: электронная и протонная. Электронная теория К. и о. Льюиса. Отличит. признаком К. и о. в теории Льюиса является то, что они взаимод. друг с другом с образованием донорно-акцепторной (координац.) связи: А+ВDА:В, где А — кислота, В — основание, А: В — кислотно-основный комплекс (продукт нейтрализации). В результате приобретенной пары электронов атомом, ответственным за кислотные свойства рассматриваемого соед., часто возникает завершенная электронная конфигурация, напр.:
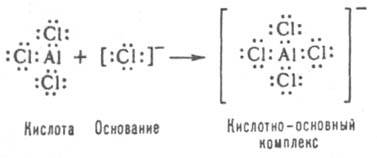
В случае взаимод. нейтральных молекул продукт реакции [напр., BF3.NH3, SbCl5.O(C2H5)2] часто называют аддуктом. К. и о. в совр. электронной теории классифицируют по типу орбиталей, принимающих участие в образовании межмол. донорно-акцепторных связей в кислотно-основном комплексе. При таком подходе все кислоты (акцепторы) разделяют на σ-, v- и π-типы, все основания (доноры) — на п-, σ- и π-типы. В образовании связи между кислотой и основанием принимает участие наиб. высокая в энергетич. отношении граничная мол. орбиталь основания и наиб. низкая орбиталь кислоты. По типу орбиталей, ответственных за их образование, донорно-акцепторные комплексы разделяют на 9 типов: nv (напр., R3N.MeXn), nσ(R3N.I2), σv(RX.МеХn), ss(RX.I2), sp(RX.ArH), πv(ArH.MeXn), ps(ArH.I2) и pp(ArH.ТЦХД), где первыми в скобках указаны доноры, вторыми — акцепторы; R — алкил, Me — металл и Х — галоген; ТЦХД — тетрацианохинодиметан. Любое основание может вступать во взаимод. с любой кислотой. Одно и то же соед. в зависимости от партнера может выступить как основание или как кислота. О специфичности кислотно-основного взаимодействия см. "жестких" и "мягких" кислот и оснований принцип. Реакции между К. и о. Льюиса иллюстрируют след, примеры:
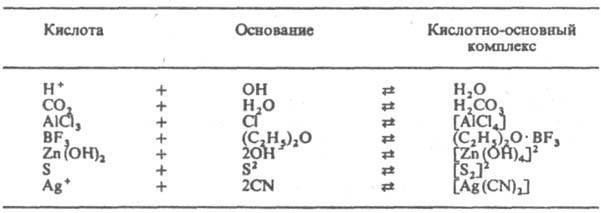
Понятие основания в теориях Льюиса и Брёнстеда совпадают, однако понятие кислоты в первой теории охватывает кроме протона также электроноакцепторные вещества, в обратимых реакциях которых с основаниями не участвует водород, напр. BF3, SO3, Ag+. К кислотам Льюиса относится протон, как частица, легко присоединяющая пару электронов. Протонные кислоты рассматриваются в теории Льюиса как продукты нейтрализации протона основаниями (напр., соляная кислота — продукт нейтрализации H+ основанием Cl− ). Растворение кислот Льюиса в ионизирующих растворителях приводит к росту концентрации катионов растворителя (напр., SO3+H2ODH3O++HSO−4). Основания же увеличивают концентрацию анионов растворителя [напр., (CH3)3N+H2ODOH−+(CH3)3NH+]. Поэтому нетрудно оттитровать К. и о. в ионизирующихся растворителях, фиксируя точку эквивалентности индикатором или электрохимически. Кислоты Льюиса можно также оттитровать в инертных растворителях, напр. удается оттитровать раствор SnCl4 в бензоле раствором (CH3)3N в этом же растворителе, используя тимоловый голубой в качестве индикатора. К. и о. Льюиса невозможно расположить в универсальный ряд по силе, т. к. их последовательность зависит от вещества, взятого за стандарт для сравнения. Однако фиксируя стандарт сравнения (оснований кислоты Льюиса располагают в ряды на основе величин изменения энтальпии DH при реакции нейтрализации, хотя использование для этих целей соответствующих величин изменения свободной энергии ΔG более строго. Стандартное вещество для определения донорной способности оснований Льюиса — SbCl5. Значение ΔH0298 реакции SbCl5 с к.-л. электронодонорным веществом наз. донорным числом (DN) данного вещества. Протонная теория К. и о. Брёнстеда . Хотя название рассматриваемой теории подчеркивает исключит. роль протона, подразумеваются все возможные ядра атома водорода: протон, дейтрон и тритон. По этой теории, кислота и основание составляют сопряженную пару и связаны уравнением: кислота D основание + протон В растворах протон не может существовать в своб. виде, он соединяется с молекулами растворителя. В воде, напр., сольватир. протон существует в виде ионов H5O2 — симметричных комплексов с сильной водородной связью [H2O...Н...OH2]+ В расчетах обычно принимают, что протон находится в воде в виде ионов гидроксония H3O+, и реакции сопряженных К. и о., которые м. б. молекулами или ионами, записывают уравнением:
АН+ВDBH++А−, (1)
где АН, ВH+ — кислоты; В, А− — основания. Первонач. вариант теории Брёнстеда рассматривал только полный переход протона от кислоты к основанию. Однако к нач. 60-х гг. было показано, что реакция между К. и о. не сводится лишь к полному переходу протона и имеет более сложный характер. Сначала при реакции между атомом водорода кислоты АН и электронодонорным атомом основания В возникает водородная связь и образуется комплекс АН... В. Во мн. случаях протолитич. реакция ограничивается этой стадией; такой процесс наз. незавершенным кислотно-основным взаимодействием. В благоприятных условиях, напр. при высокой диэлектрич. проницаемости растворителя ε, происходит передача протона от кислоты к основанию, в результате чего основание протонируется (завершенное кислотно-основное взаимод.). Образовавшиеся ионы могут находиться в растворе в виде ионных пар или в своб. виде. Весь кислотно — основной процесс м. б. выражен схемой:
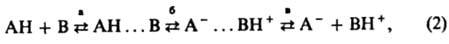
где стадии а и б — соотв. незавершенное и завершенное кислотно-основное взаимод., стадия в диссоциация ионной пары на своб. ионы. Согласно этой схеме, А. И. Шатенштейном в 1960 предложены след, определения, соответствующие совр. состоянию протонной теории К. и о.: основание — электронодонорный реагент, обладающий сродством к протону, кислота — электроноакцепторный реагент, в равновесных реакциях которого с основанием участвует водород. Кислота соединяется с основанием в результате образования между ними водородной связи или отдает ему протон. Во мн. случаях схема кислотно-основного процесса упрощается, напр., в водной среде (ε H2O=78,5), как правило, она сводится к уравнению (1). Уравнение реакции кислоты АН с водой имеет вид:
АН+H2ODА+H3O+ (3)
В этом уравнении не учтено, что протон существует в виде иона H5О+2 и для реакции необходимы две молекулы H2O. Константа равновесия реакции (3) выражается соотношением:

где аА-, аH3O+, аAH и аН2O — термодинамич. активности соответствующих частиц. Кислотность разных кислот можно измерить лишь относительно к.-л. произвольно выбранной пары сопряженных К. и о. Обычно в качестве последней используют пару H3O+, H2O. Поскольку в разб. растворах количество растворителя величина практически постоянная, константу соответствующего равновесия К=aH+∙аН2O/aH3O+ (aH+ — активность ионов H+) приравнивают к единице, что приводит к отношению аH+=аН3O+/аH2O. В рамках сделанного допущения константа кислотности кислоты Ka имеет вид:

Аналогично протону ион OH− в растворах сольватирован; в воде он существует в виде ионов H3O2− — симметричных комплексов с сильной водородной связью [НО...Н...ОН]−. В нижеприведенных формулах сольватация OH− не учитывается и реакция основания В с водой описывается уравнением:
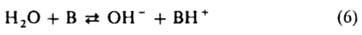
Константа равновесия этой реакции:

Основание В характеризуют константой основности:

или константой кислотности его сопряженной кислоты:
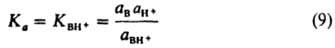
В амфотерных растворителях SН происходит автопротолиз, т. е. реакция, где одна молекула растворителя ведет себя как кислота относительно другой такой же молекулы, выполняющей роль основания:
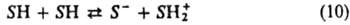
Поскольку по уравнению (10) реагирует небольшая доля растворителя SН, то в качестве постоянной рассматривают константу автопротолиза (ионное произведение) растворителя SH:
KS=aS-aSH+2, (11)
которая связана с Kа и Kb соотношением:
KS=Ka∙Kb (12)
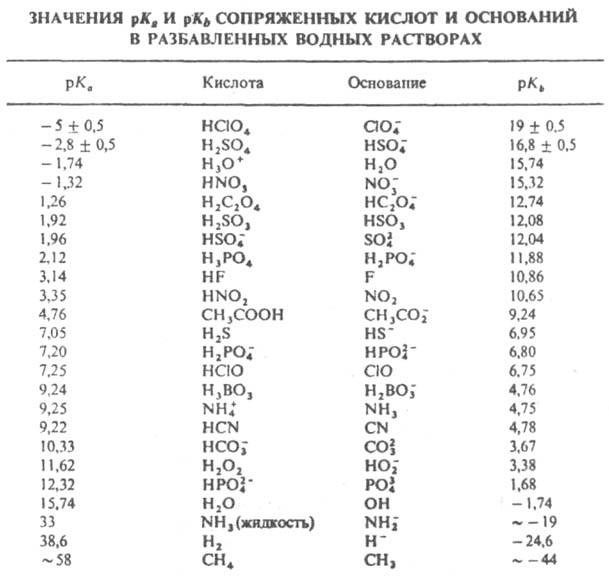
Согласно этому уравнению, кислота тем сильнее, чем слабее сопряженное основание, и наоборот. Вместо величин Kа и Kb по аналогии с водородным показателем pH чаще используют соответствующие значения рК=-lgK. Из уравнения (12) для водных растворов следует, что pKH2O=pKа+pKb, или pKа=14-pKb,. Кислоты можно разделить на очень сильные (р/Ka<0), сильные (0а<4,5), средней силы (4,5а<9), слабые (9 а<14), очень слабые (pKа>14); см. табл. В выражениях (4), (5), (7) — (9) не учитывается существование протона в виде H5O+2 и гидроксила в виде H3O−2. Однако это не сказывается на величинах констант, т. к. активность чистого растворителя принимается равной 1. В протонной теории К. и о. понятия кислоты и основания относятся лишь к функции, которую выполняет рассматриваемое соед. в протолитич. реакции. Одно и то же соед. может в одних условиях реагировать как кислота, а в других-как основание. Например, в водном растворе CH3COOH ведет себя как кислота, а в среде 100%-ной H2SO4 как основание. Большое влияние на кислотно-основное взаимод. оказывает растворитель, в среде которого происходит рассматриваемый процесс. Добавленная к растворителю М кислота АН дает ассоциат с растворителем, в котором происходит перераспределение электронной плотности с образованием связи близкой к ионной; затем осуществляется диссоциация:
Подразумевается, что сольватированы как ионная пара, так и своб. ионы. Сольватация реагентов и продуктов реакции значительно влияет на относит. силу кислот. Существуют методы исследования равновесий между протоном и основаниями в газовой фазе, напр. масс-спектрометрия высокого давления и ион-циклотронный резонанс, где отсутствуют эффекты сольватации. На основании результатов указанных исследований составлена шкала сродства к протону-энергетич. эффекта протонизации одного моля оснований в газовой фазе. В свете электронной теории К. и о. вещества, рассматриваемые как кислоты, протонной теорией не выделяются среди прочих. Однако с учетом научной традиции и специфич. свойств протона понятие кислоты обычно используют применительно к реагентам, отщепляющим ион водорода, см., напр., карбоновые кислоты, CH-Кислоты. Кислоты неорганические. Когда пишут о кислотно-основном взаимод. веществ, не содержащих протонов, то такие вещества наз. апротонными кислотами, льюисовскими кислотами, кислотоподобными веществами, антиоснованиями или просто акцепторами. Понятия К. и о. оказывают разностороннее влияние на формирование и совершенствование мн. теоретич. концепций во всех осн. хим. дисциплинах. Это свидетельствует о чрезвычайно широкой распространенности в природе процессов, связанных с кислотно-основными взаимодействиями. Из всех теорий К. и о. протонной теории удалось создать наиб. разработанный количеств. подход к рассматриваемым явлениям. На основании этой теории разработаны такие разделы хим. наук, как pH-метрия в неводных средах, гомог. кислотно-основной катализ, теория функций кислотности и др.
Лит.: Шатенштейн А. И., Теории кислот и оснований, М.-Л., 1949; его же, Изотопный обмен и замещение водорода в органических соединениях, в свете теоряи кислот и основании, М., 1960; Либрович Н. Б., Майоров В. Д.. Савельев В. А.. Докл. АН СССР, 1975, т. 225, М 6, с. 1358–1361; Белл Р Д., Протон в химии, пер. с англ., М., 1977; Мискиджьан С П Гарновскни А. Д., Введение в современную теорию кислот и оснований. К., 1979; Кабачник М.И., "Успехи химии". 1979, т. 48, в. 9, с 1523 47, The international encyclopedia of physical chemistry and chemical physics. Topic IS. v. 4-Acid-base equilibna Oxf.-L-N.Y, 1945; Jensen W В., The Lewis arid-base concepts. An overview. N.Y4 1980.
Ю. Л. Халдна
Значения в других словарях
- Кислоты и основания — Классы химических соединений. Обычно кислотами называют вещества, содержащие водород (HCl, HNO3, H2SO4, CH3COOH и т.д.) и диссоциирующие в воде с образованием ионов Н+ (точнее, ионов гидроксония H3O+). Большая советская энциклопедия
