кулонометрия
КУЛОНОМЕТРИЯ
электрохим метод исследования и анализа, основанный на измерении количества электричества (Q), прошедшего через электролизер при электрохим. окислении или восстановлении вещества на рабочем электроде. Согласно объединенному Фарадея закону, масса электрохимически превращенного вещества (Р) в г связана с Q в Кл соотношением: Р=QM/Fn, где М-молекулярная или атомная масса вещества, п- число электронов, вовлеченных в электрохим. превращение одной молекулы (атома) вещества (М/n — электрохимический эквивалент вещества), F — постоянная Фарадея. К. — единственный физ.-хим. метод анализа, в котором не требуются стандартные образцы. Различают прямую К. и кулонометрич. титрование (К. т.). В первом случае определяют электрохимически активное вещество, во втором случае — независимо от электрохим. активности определяемого вещества в испытуемый раствор вводят электрохимически активный вспомогат. реагент, продукт электрохим. превращения которого (кулонометрич. титрант) с большой скоростью и количественно химически взаимодействует с определяемым веществом. Оба варианта К. можно проводить при постоянном потенциале Е рабочего электрода (потенциостатич. режим) или при постоянном токе электролиза Iэ (гальваностатич. режим). Наиб. часто используются прямая К. при постоянном Е и К.т. при постоянном Iэ. Для кулонометрич. анализа необходимо соблюдение след. условий: электрохим. превращение вещества должно протекать со 100%-ным выходом по току (η), т. е. должны отсутствовать побочные электрохим. и хим. процессы; нужны надежные способы определения количества электричества и установления момента завершения электрохим. или хим. реакции. В прямой К. 100%-ный выход по току обеспечивается, если значение Е поддерживать постоянным в области предельного диффузионного тока Iпp на вольтамперограмме определяемого вещества (см. вольтамперометрия). При этом в анализируемом растворе должны отсутствовать посторонние вещества, способные электрохимически превращ. в тех же условиях. Количество электричества определяют обычно с помощью электронных интеграторов тока. Иногда пользуются менее точными приборами — кулонометрами разл. типа, а также планометрическим и расчетными методами. В последних двух случаях завершением электролиза считают момент, когда Iэ падает до значения фонового тока Iф, поэтому количество электричества, необходимое для завершения электродной реакции, равно разности Qоб—Qф, где Qоб — общее количество электричества, Qф — количество электричества, измеренное в тех же условиях за то же время электролиза tэ, но в отсутствие определяемого вещества. Если электрохим. реакция первого порядка, то It=I0е−Kt=I0∙10−Kt, К=2,30ЗК'=SD/Vd, где It и I0 — ток электролиза соответственно в момент времени т и при t=0, S — площадь поверхности электрода, D — коэф. диффузии электрохимически активного вещества, d — толщина диффузионного слоя, V — объем раствора в ячейке. Продолжительность электролиза не зависит от начальной концентрации вещества, но заметно сокращается с увеличением соотношения S/V и при интенсивном перемешивании раствора (уменьшении 6). Можно считать электролиз завершенным, когда Iэ станет равен 0,1 I0 или 0,01 I0 (в зависимости от требуемой точности анализа). В планометрич. способе для установления Q измеряют площадь под кривой It — t, т. к. 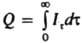 . В расчетном способе решают последнее уравнение, подставляя в него выражение для It. После интегрирования получают Q=I0/К=I0/2,303K'. Для нахождения I0 и К' выражение для It логарифмируют и по нескольким (5–7) точкам строят прямую lgIt-t, тангенс угла наклона которой равен К', а точка пересечения с осью ординат соответствует lgI0, т. е. для определения Q нет необходимости проводить электролиз до конца и измерять I0, значение которого плохо воспроизводится. В др. расчетном способе Q вычисляют по формуле Q=[Q22-Q1Q3]/(2Q2-Q1-Q3), где Q1, Q2 и Q3 — количества электричества в моменты времени t1, t2 и t3 соотв., причем (t2-t1)~(t3-t2). Прямая К. при постоянном Iэ осуществима, только когда определяемое вещество находится или предварительно выделено в виде твердой фазы на рабочем электроде. Анодное (катодное) растворение при постоянном Iэ и измерение tэ в момент резкого изменения Е позволяет рассчитать Q=Iэtэ. Преимущество прямой К. перед К.т. — высокая селективность. Однако наиболее распространенный метод кулонометрич. анализа — К. т. в гальваностатич. режиме, т. к. он отличается простотой аппаратурного оформления и более высокой точностью. Для нахождения оптим. условий проведения эксперимента вычисляют значения выхода по току по формуле η=(iэ-iф)∙100/iэ, где iэ и iф — плотности тока электролиза (т. е. отношения Iэ/S) соотв. в присутствии вспомогат. реагента и без него при одних и тех же значениях Е. Варьируя температуру, pH среды, концентрации электрохимически активного вещества и разл. фоновых электролитов, а также значения iэ, строят графики h=f(iэ) и находят область iэ, при которой h~100%. Существуют и другие, реже используемые способы расчета оптим. значения iэ. Обычно вычисляют также эффективность титрования Qэ∙100/Qt (%), где Q, и (Qt — соотв. эксперим. и тeоретич. значения количества электричества при К. т. известной массы определяемого вещества. Если определяемое вещество А электрохимически активно, его предельный ток должен быть меньше тока электролиза и значительно меньше предельного тока (I'пр) вспомогат. реагента С. При этом в электролизере происходят электрохим. реакции: A6ne:В и Сbme:D, а также химическая реакция (окисление-восстановление, комплексообраювание, осаждение или кислотно-основное взаимод.) mA+nD:mB+nС или mА+nD:AmDn. При электролизе концентрация реагента С остается постоянной (если он регенерируется) или меняется незначительно, т. к. его концентрация в растворе на 3–4 порядка превышает концентрацию определяемого вещества. Таким образом, значение I'пр практически постоянно. Поэтому вещество С называют электрохим. буфером, поддерживающим постоянное значение Е. В К. т. время электролиза мало, т. к. содержание А в электролизере уменьшается одновременно вследствие электрохим. и хим. реакций. Если А неэлектроактивно, то для выбора оптим. значения iэ предварительно определяют зависимость выхода по току вещества D от iэ, как описано выше. Конец хим. реакции устанавливают с помощью цветных индикаторов или физ. — хим. методами. Среди последних наиб. удобны потенциометры и амперометрия с одним или двумя поляризованными электродами (см. амперометрическое титрование). Количество электричества рассчитывают по формуле Q=Iэtэ. Кулонометрич. титрант получают из растворимых солей, твердых электрохимически активных материалов (Ag, Hg), амальгам, электродов второго рода и из воды (при определении кислот и оснований) в присутствии инертных электролитов, создающих необходимую электропроводность раствора. Преимущества К. т. перед обычными титриметрич. методами: нет необходимости стандартизовать растворы титранта; титрант прибавляется очень малыми порциями (практически непрерывно); раствор не разбавляется; можно генерировать электрохимически неактивные титранты, напр. комплексон III, а также малоустойчивые сильные окислители и восстановители, в частности Mn(III), Pb(IV), Cr(П), V(II), Ti(III). Установки для кулонометрич. анализа (рис. 1,2) состоят из потенциостата или гальваностата, регистрирующего потенциометра или интегратора тока, электролизера и индикац. системы (в случае использования физ.-хим. методов для
. В расчетном способе решают последнее уравнение, подставляя в него выражение для It. После интегрирования получают Q=I0/К=I0/2,303K'. Для нахождения I0 и К' выражение для It логарифмируют и по нескольким (5–7) точкам строят прямую lgIt-t, тангенс угла наклона которой равен К', а точка пересечения с осью ординат соответствует lgI0, т. е. для определения Q нет необходимости проводить электролиз до конца и измерять I0, значение которого плохо воспроизводится. В др. расчетном способе Q вычисляют по формуле Q=[Q22-Q1Q3]/(2Q2-Q1-Q3), где Q1, Q2 и Q3 — количества электричества в моменты времени t1, t2 и t3 соотв., причем (t2-t1)~(t3-t2). Прямая К. при постоянном Iэ осуществима, только когда определяемое вещество находится или предварительно выделено в виде твердой фазы на рабочем электроде. Анодное (катодное) растворение при постоянном Iэ и измерение tэ в момент резкого изменения Е позволяет рассчитать Q=Iэtэ. Преимущество прямой К. перед К.т. — высокая селективность. Однако наиболее распространенный метод кулонометрич. анализа — К. т. в гальваностатич. режиме, т. к. он отличается простотой аппаратурного оформления и более высокой точностью. Для нахождения оптим. условий проведения эксперимента вычисляют значения выхода по току по формуле η=(iэ-iф)∙100/iэ, где iэ и iф — плотности тока электролиза (т. е. отношения Iэ/S) соотв. в присутствии вспомогат. реагента и без него при одних и тех же значениях Е. Варьируя температуру, pH среды, концентрации электрохимически активного вещества и разл. фоновых электролитов, а также значения iэ, строят графики h=f(iэ) и находят область iэ, при которой h~100%. Существуют и другие, реже используемые способы расчета оптим. значения iэ. Обычно вычисляют также эффективность титрования Qэ∙100/Qt (%), где Q, и (Qt — соотв. эксперим. и тeоретич. значения количества электричества при К. т. известной массы определяемого вещества. Если определяемое вещество А электрохимически активно, его предельный ток должен быть меньше тока электролиза и значительно меньше предельного тока (I'пр) вспомогат. реагента С. При этом в электролизере происходят электрохим. реакции: A6ne:В и Сbme:D, а также химическая реакция (окисление-восстановление, комплексообраювание, осаждение или кислотно-основное взаимод.) mA+nD:mB+nС или mА+nD:AmDn. При электролизе концентрация реагента С остается постоянной (если он регенерируется) или меняется незначительно, т. к. его концентрация в растворе на 3–4 порядка превышает концентрацию определяемого вещества. Таким образом, значение I'пр практически постоянно. Поэтому вещество С называют электрохим. буфером, поддерживающим постоянное значение Е. В К. т. время электролиза мало, т. к. содержание А в электролизере уменьшается одновременно вследствие электрохим. и хим. реакций. Если А неэлектроактивно, то для выбора оптим. значения iэ предварительно определяют зависимость выхода по току вещества D от iэ, как описано выше. Конец хим. реакции устанавливают с помощью цветных индикаторов или физ. — хим. методами. Среди последних наиб. удобны потенциометры и амперометрия с одним или двумя поляризованными электродами (см. амперометрическое титрование). Количество электричества рассчитывают по формуле Q=Iэtэ. Кулонометрич. титрант получают из растворимых солей, твердых электрохимически активных материалов (Ag, Hg), амальгам, электродов второго рода и из воды (при определении кислот и оснований) в присутствии инертных электролитов, создающих необходимую электропроводность раствора. Преимущества К. т. перед обычными титриметрич. методами: нет необходимости стандартизовать растворы титранта; титрант прибавляется очень малыми порциями (практически непрерывно); раствор не разбавляется; можно генерировать электрохимически неактивные титранты, напр. комплексон III, а также малоустойчивые сильные окислители и восстановители, в частности Mn(III), Pb(IV), Cr(П), V(II), Ti(III). Установки для кулонометрич. анализа (рис. 1,2) состоят из потенциостата или гальваностата, регистрирующего потенциометра или интегратора тока, электролизера и индикац. системы (в случае использования физ.-хим. методов для
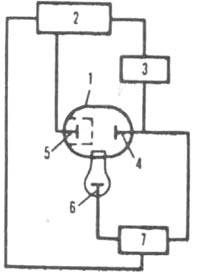
Рис. 1. Схема установки для прямой кулонометрии при постоянном E: 1 электролизер; 2 источник постоянного токa с регулируемым напряжением: 3 прибор для определения количества злектричества: 4 рабочий электрод; 5 вспомогательный электрод; 6 электрод сравнения, относительно которого контролируют потенциал рабочего электрода: 7 устройство, измеряющее разность потенциалов.
установления конца хим. реакции в К. т.). Приборы для К. легко автоматизируются. Электролизеры (см., напр., на рис. 2) представляют собой, как правило, стеклянные сосуды, катодные и анодные камеры в которых разделены диафрагмой (напр., из пористого стекла). В качестве рабочих и вспомогательных (замыкающих цепь электролиза) электродов используют благородные металлы (Pt, Au), электроды второго рода и, реже, углеродные материалы (графит, стеклоуглерод и др.). Раствор, в который погружен рабочий электрод, перемешивают обычно магн. мешалкой; при необходимости эксперимент проводят в атмосфере инертного газа. К. применяют для определения как следовых, так и весьма больший количеств веществ с высокой точностью. Погрешность прямой К. в потенциостатич. режиме обычно 0,5–1%, а К.т. в гальваностатич. режиме — 0,1–0,3%. Особенно точен дифференциальный вариант К. В этом случае в цепь последовательно включают два идентичных электролизера, в один из которых вносят стандартное вещество в известном количестве, эквивалентном количеству электричества Q1, которое на величину Q2 меньше количества электричества, необходимого для завершения электрохим. или хим. реакции определяемого вещества во втором электролизере. Электролиз проводят в одинаковых условиях при строгом контроле значений Е и Iэ. Все погрешности сказываются только на количестве электричества Q2, которое обычно
Рис. 2. Схема установки для кулонометрич. титрования: 1 электролизер: 2 рабочий электрод (электрод генерации): 3 — вспомогательный электрод: 4 пористое стекло: 5 прецизионное сопротивление: 6 устройство, измеряющее разность потенциалов: 7 источник постоянного тока: 8 хронометр: 9 магнитная мешалка.
составляет 2–5% Q1. Содержание определяемого вещества соответствует сумме Q1+Q2. Чувствительность кулонометрич. методов определяется в осн. способами установления момента завершения электрохим. или хим. реакции и составляет 10−8–10−9 моль/л. Использование неводных и водно-орг. сред расширяет область потенциалов, в которой протекают электрохим. и хим. реакции, и таким образом увеличивает круг веществ, анализируемых кулонометрически. К. применяют для анализа мн. неорг. (практически все металлы, галогены, S и др.) и орг. веществ (ароматич. амины, нитро- и нитрозосоединения, фенолы, азокрасители, алифатич. амиды и др.); определения воды в орг. веществах; установления толщины и анализа металлич. покрытий; изучения процессов коррозии; исследования кинетики и механизма хим. реакций (в т. ч. каталитических); определения констант равновесия реакций; установления числа электронов, участвующих в электрохим. и хим. взаимодействиях, и т. д. Кулонометрич. детекторы широко используются в проточно-инжекционном анализе и хроматографии (см. детекторы хроматографические).
Лит.: Зозуля А. Н.. Кулонометрический анализ, 2 изд., Л.. 1968: Агасян П. К., Хамракулов Т. К.. Кулонометрический метод анализа. М., 1984.
П. К. Агасян, Л. Б. Оганесян
Значения в других словарях
- Кулонометрия — Один из электрохимических методов анализа (См. Электрохимические методы анализа). Большая советская энциклопедия
- кулонометрия — орф. кулонометрия, -и Орфографический словарь Лопатина
- кулонометрия — КУЛОНОМЕТРИЯ и, ж. Совокупность электро-химических методов анализа, основанных на измерении количества электричества, расходуемого при выделении на электроде того или иного вещества. Словарь галлицизмов русского языка
- Кулонометрия — (a. coulombmetry; н. Coulombometrie; ф. coulombmetrie; и. culombiometria) — электрохим. метод исследования и анализа веществ, основанный на измерении кол-ва электричества, расходуемого на электролитич. восстановление или окисление вещества. B основу K. Горная энциклопедия
- КУЛОНОМЕТРИЯ — КУЛОНОМЕТРИЯ — совокупность электрохимических методов анализа, основанных на измерении количества электричества, расходуемого при выделении на электроде того или иного вещества. Применяется для установления толщины металлических покрытий, оксидных пленок; в химическом анализе. Большой энциклопедический словарь
