цветность органических соединений
ЦВЕТНОСТЬ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
зависимость цвета орг. соед. от их строения. В статье рассмотрены основные положения электронной теории цветности. Ощущение цвета возникает в результате воздействия на зрительный нерв электромагн. излучения с частотами v в пределах 3,8∙10|4-7,6∙1014 Гц, т. е. с длинами волн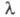 380–760 нм (т. наз. видимая часть спектра). Суммарное действие электромагн. излучений во всем указанном интервале вызывает ощущение белого цвета, отсутствие определенного интервала длин волн — окрашенного (см. цветометрия). В табл. 1 приведены приблизит, границы интервалов длин волн монохро-матич. световых лучей (т. наз. спектральные цвета) и дополнит. цвета, которые возникают в зрительном аппарате, если из белого луча изымается (поглощается) к.-л. из спектральных цветов.
380–760 нм (т. наз. видимая часть спектра). Суммарное действие электромагн. излучений во всем указанном интервале вызывает ощущение белого цвета, отсутствие определенного интервала длин волн — окрашенного (см. цветометрия). В табл. 1 приведены приблизит, границы интервалов длин волн монохро-матич. световых лучей (т. наз. спектральные цвета) и дополнит. цвета, которые возникают в зрительном аппарате, если из белого луча изымается (поглощается) к.-л. из спектральных цветов.
Табл. 1 — ПРИМЕРНЫЕ ГРАНИЦЫ ОСНОВНЫХ ЦВЕТОВ СПЕКТРА
таблица в процессе добавления
* Голубой.
Белое тело практически полностью отражает лучи всей видимой части спектра, черное — полностью поглощает их, серое — поглощает все лучи приблизительно одинаково, но не полностью, цветное — избирательно поглощает некоторые из них.
Энергия Е электромагн. излучения определяется уравнением Планка: 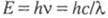 (h — постоянная Планка; с — скорость света;
(h — постоянная Планка; с — скорость света; 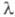 - длина волны) и составляет для видимой части спектра ~ 158–300 кДж/моль. Для того чтобы соед. было окрашенным, энергия возбуждения его молекулы
- длина волны) и составляет для видимой части спектра ~ 158–300 кДж/моль. Для того чтобы соед. было окрашенным, энергия возбуждения его молекулы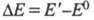 (Е0 и E' — энергия молекулы соотв. в основном и возбужденном состояниях) должна лежать в этих пределах (при
(Е0 и E' — энергия молекулы соотв. в основном и возбужденном состояниях) должна лежать в этих пределах (при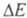 > 300 кДж/моль поглощение происходит в УФ, при
> 300 кДж/моль поглощение происходит в УФ, при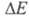 < 158 кДж/моль — в ИК частях спектра). Указанным значениям энергии возбуждения отвечают переходы между разл. электронными уровнями энергии молекул (см. молекулярные спектры).
< 158 кДж/моль — в ИК частях спектра). Указанным значениям энергии возбуждения отвечают переходы между разл. электронными уровнями энергии молекул (см. молекулярные спектры).
Поглощение света веществом характеризуется кривой поглощения, которая строится на основе измерения интенсивностей поглощения света определенных длин волн, рассчитанных по закону Бугера — Ламберта — Бера: 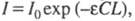 где I0 и I — интенсивности светового луча соотв. до и после прохождения через раствор вещества; С — молярная концентрация вещества; L — толщина слоя раствора;
где I0 и I — интенсивности светового луча соотв. до и после прохождения через раствор вещества; С — молярная концентрация вещества; L — толщина слоя раствора; 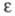 - молярный коэф. поглощения, или экстинкции, характерный для каждого вещества. Если кривая поглощения построена в координатах
- молярный коэф. поглощения, или экстинкции, характерный для каждого вещества. Если кривая поглощения построена в координатах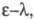 то положение ее максимума на оси абсцисс
то положение ее максимума на оси абсцисс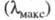 характеризует спектральный цвет и является мерой энергии возбуждения, а положение максимума на оси ординат
характеризует спектральный цвет и является мерой энергии возбуждения, а положение максимума на оси ординат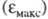 — интенсивность окраски и является мерой вероятности электронного перехода (рис. 1).
— интенсивность окраски и является мерой вероятности электронного перехода (рис. 1).
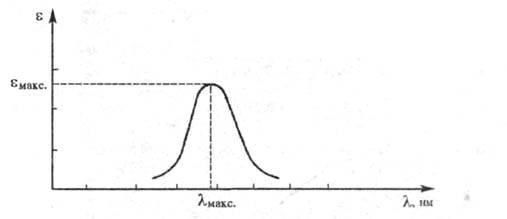
Рис. 1. Спектральная кривая поглощения.
С уменьшением энергии возбуждения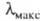 смещается в длинноволновую часть спектра, при этом окраска изменяется от желтой к оранжевой, красной и т. д.; такое изменение цвета наз. его углублением или батохромным сдвигом; увеличение энергии возбуждения, приводящее к смещению
смещается в длинноволновую часть спектра, при этом окраска изменяется от желтой к оранжевой, красной и т. д.; такое изменение цвета наз. его углублением или батохромным сдвигом; увеличение энергии возбуждения, приводящее к смещению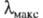 в коротковолновую область и изменению окраски в обратной последовательности, наз. повышением цвета или гипсохромным сдвигом.
в коротковолновую область и изменению окраски в обратной последовательности, наз. повышением цвета или гипсохромным сдвигом.
Первостепенное значение в процессах поглощения света молекулами орг. соед. имеет разность энергий их граничных мол. орбиталей (ГМО) — высшей занятой и низшей свободной, т. к. переход электронов с одной орбитали на другую обычно обусловливает длинноволновую полосу поглощения, лежащую в видимой части спектра и определяющую цвет соед. Уровни ГМО зависят от характера электронов, входящих в состав молекулы.
Для H2 и парафинов, содержащих только связи С — Н и С — С, энергия возбуждения для перехода
связи С — Н и С — С, энергия возбуждения для перехода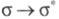 составляет (кДж/моль): 1090 (H2), 1000 (CH4), 890 (C2H6), что соответствует поглощению в дальней УФ области (
составляет (кДж/моль): 1090 (H2), 1000 (CH4), 890 (C2H6), что соответствует поглощению в дальней УФ области (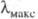 110, 120 и 135 нм соотв.). Такие соед. бесцветны.
110, 120 и 135 нм соотв.). Такие соед. бесцветны.
Для молекул углеводородов с изолир.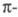 связями появляется возможность
связями появляется возможность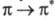 — перехода; при этом энергия возбуждения снижается и составляет, напр., для этилена 739 кДж/моль, что соответствует поглощению в дальней УФ области (
— перехода; при этом энергия возбуждения снижается и составляет, напр., для этилена 739 кДж/моль, что соответствует поглощению в дальней УФ области ( 162,5 нм). Такие соед. также бесцветны.
162,5 нм). Такие соед. также бесцветны.
Иное явление наблюдается для углеводородов с сопряженными двойными связями, у которых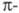 электроны делокализованы. С увеличением углеводородной цепи уровни энергии ГМО сопряженных
электроны делокализованы. С увеличением углеводородной цепи уровни энергии ГМО сопряженных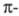 связей расщепляются и появляются новые уровни, переход между которыми требует меньших затрат энергии по сравнению с этиленом. Так, для бутадиена энергии
связей расщепляются и появляются новые уровни, переход между которыми требует меньших затрат энергии по сравнению с этиленом. Так, для бутадиена энергии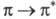 -перехода 553 кДж/моль, что соответствует поглощению при
-перехода 553 кДж/моль, что соответствует поглощению при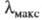 217 нм (т. е. в ближнем ультрафиолете). Одновременно значительно возрастает интенсивность полос поглощения. По мере удлинения сопряженной цепочки происходит дальнейшее сближение уровней ГМО, в результате чего имеет место систематич. смещение полосы поглощения в длинноволновую часть спектра и появление окраски.
217 нм (т. е. в ближнем ультрафиолете). Одновременно значительно возрастает интенсивность полос поглощения. По мере удлинения сопряженной цепочки происходит дальнейшее сближение уровней ГМО, в результате чего имеет место систематич. смещение полосы поглощения в длинноволновую часть спектра и появление окраски.
Аналогичное действие оказывает увеличение замкнутой (ароматической) системы сопряженных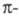 связей, особенно если ее отдельные звенья расположены линейно, что обеспечивает возможность делокализации
связей, особенно если ее отдельные звенья расположены линейно, что обеспечивает возможность делокализации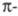 электронов (табл. 2).
электронов (табл. 2).
Смещение полосы поглощения в длинноволновую часть спектра происходит также при наличии в сопряженной системе как электронодонорных, так и электроноакцепторных заместителей, которые усиливают делокализацию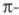 электронов в основном состоянии, что приводит к сближению их ГМО и углублению цвета (табл. 3).
электронов в основном состоянии, что приводит к сближению их ГМО и углублению цвета (табл. 3).
Важным фактором влияния электронодонорных и электроноакцепторных заместителей на электронный спектр поглощения является снятие запретов по симметрии на вероятность электронных переходов. Например, у бензола первые два длинноволновых электронных перехода запрещены по симметрии.
Табл. 2 — ЭНЕРГИИ ВОЗБУЖДЕНИЯ И ДЛИННОВОЛНОВЫЕ ПОЛОСЫ ПОГЛОЩЕНИЯ АЛИФАТИЧЕСКИХ И АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
таблица в процессе добавления
Табл. 3 — ДЛИННОВОЛНОВЫЕ ПОЛОСЫ ПОГЛОЩЕНИЯ РАЗЛИЧНЫХ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
таблица в процессе добавления
Введение заместителей снимает запрет и увеличивает интенсивность поглощения света. Так, молярный коэф. поглощения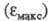 фенола в ~ 7, анилина в 8, нитробензола в 45, 4-нитрофенола в 56,4-нитроанилина в 72 раза больше
фенола в ~ 7, анилина в 8, нитробензола в 45, 4-нитрофенола в 56,4-нитроанилина в 72 раза больше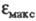 бензола (для длинноволновых максимумов).
бензола (для длинноволновых максимумов).
Влияние заместителей м. б. усилено или ослаблено ионизацией. Так, в кислой среде усиливаются электроноакцепторные свойства карбонильной группы в результате присоединения протона и появления эффективного положительного заряда
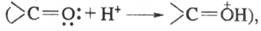 электронодонорные свойства аминогруппы вследствие перехода азота в новое валентное состояние исчезают
электронодонорные свойства аминогруппы вследствие перехода азота в новое валентное состояние исчезают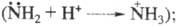 в щелочной среде усиливаются электронодонорные свойства гидроксигруппы благодаря тому, что кислород приобретает эффективный отрицательный заряд
в щелочной среде усиливаются электронодонорные свойства гидроксигруппы благодаря тому, что кислород приобретает эффективный отрицательный заряд Эти изменения отражаются на поглощении света соединениями (табл. 4).
Эти изменения отражаются на поглощении света соединениями (табл. 4).
Табл. 4 — ДЛИННОВОЛНОВЫЕ ПОЛОСЫ ПОГЛОЩЕНИЯ ИОНИЗИРОВАННЫХ МОЛЕКУЛ
таблица в процессе добавления
Усиление электронодонорных и электроноакцепторных свойств одновременно с углублением цвета увеличивает и интенсивность поглощения. Для 4-нитрофенолят-аниона в ~ 1,9 раза превосходит
в ~ 1,9 раза превосходит 4-нитрофенола. Напротив, утрата электронодонорных свойств аминогруппы в результате ионизации в кислой среде приводит к уменьшению интенсивности поглощения: если
4-нитрофенола. Напротив, утрата электронодонорных свойств аминогруппы в результате ионизации в кислой среде приводит к уменьшению интенсивности поглощения: если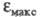 анилина в 8 раз больше, чем
анилина в 8 раз больше, чем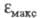 бензола, то анилиний-катион поглощает практически с той же интенсивностью, что и бензол.
бензола, то анилиний-катион поглощает практически с той же интенсивностью, что и бензол.
Цветность орг. соед. связана с их принадлежностью к альтернантным или неальтернантным системам (см. альтернантные углеводороды). Так, введение в молекулу альтернантного углеводорода электронодонорного заместителя (напр., в положение 1 или 2 нафталина) всегда вызывает батохромный сдвиг полосы поглощения, для неальтернантных углеводородов эта закономерность не соблюдается; напр., введение группы CH3 в молекулу азулена ( 580 нм) может привести к батохромному сдвигу (в положение 1 или 3 — до 608 нм, а в 5 или 7 — до 592 нм) либо к гипсохромному (в положение 2 — до 566 нм, 4 или 8 — до 568 нм, а в 6 — до 565 нм).
580 нм) может привести к батохромному сдвигу (в положение 1 или 3 — до 608 нм, а в 5 или 7 — до 592 нм) либо к гипсохромному (в положение 2 — до 566 нм, 4 или 8 — до 568 нм, а в 6 — до 565 нм).
Имеет значение также принадлежность орг. соед. к четным или нечетным альтернантным системам. Четные альтернантные углеводороды в основном состоянии содержат четное число электронов, заполняющих попарно все связывающие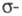 и
и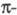 молекулярные орбитали (МО), энергия которых меньше энергии орбиталей атомов, входящих в состав молекулы. Молекулы нечетных альтернантных углеводородов в электрически нейтральном состоянии (радикалы) содержат нечетное число электронов, заполняющих попарно все связывающие МО, и один неспаренный электрон, который находится на т. наз. несвязывающей орбитали (НМО), последняя по своей природе является
молекулярные орбитали (МО), энергия которых меньше энергии орбиталей атомов, входящих в состав молекулы. Молекулы нечетных альтернантных углеводородов в электрически нейтральном состоянии (радикалы) содержат нечетное число электронов, заполняющих попарно все связывающие МО, и один неспаренный электрон, который находится на т. наз. несвязывающей орбитали (НМО), последняя по своей природе является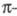 орбиталью. Если частица представляет собой мол. анион, на НМО находится электронная пара, если же частица представляет собой мол. катион, эта орбиталь остается вакантной. В приближении линейной комбинации атомных орбиталей (см. ЛКАО-приближение) коэф. при орбиталях всех непомеченных атомов равны нулю, а для помеченных (метят только нечетные атомы) — имеют конечные значения, т. е. для электронов, находящихся на НМО, электронная плотность сосредоточена только на помеченных атомах, тогда как на низшей свободной МО (НСМО) она распределена равномерно по всем помеченным и непомеченным атомам.
орбиталью. Если частица представляет собой мол. анион, на НМО находится электронная пара, если же частица представляет собой мол. катион, эта орбиталь остается вакантной. В приближении линейной комбинации атомных орбиталей (см. ЛКАО-приближение) коэф. при орбиталях всех непомеченных атомов равны нулю, а для помеченных (метят только нечетные атомы) — имеют конечные значения, т. е. для электронов, находящихся на НМО, электронная плотность сосредоточена только на помеченных атомах, тогда как на низшей свободной МО (НСМО) она распределена равномерно по всем помеченным и непомеченным атомам.
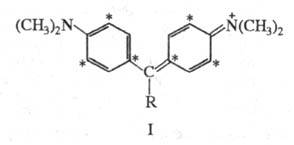
Эти особенности НМО проявляются при введении в молекулы ароматич. соед. гетероатомов. Так, если к центральному атому С (непомеченному) молекулы синего гидрола Михлера (формула I; R = H, 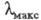 603,5 нм) присоединяется группа NH2, энергия НМО не изменяется, энергия же НСМО повышается, в результате увеличивается энергия возбуждения (
603,5 нм) присоединяется группа NH2, энергия НМО не изменяется, энергия же НСМО повышается, в результате увеличивается энергия возбуждения (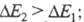 рис. 2) и полоса поглощения смещается гипсохромно до
рис. 2) и полоса поглощения смещается гипсохромно до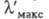 420 нм (I; R= NH2; желтый аурамин).
420 нм (I; R= NH2; желтый аурамин).
Одновременно появляется новая n-орбиталь, на которой размещается неподеленная электронная пара атома группы NH2' ee энергия ниже энергии НМО вследствие большей электроотрицательности атома N по сравнению с атомом С и возникает переход, обусловливающий появление новой полосы поглощения с
переход, обусловливающий появление новой полосы поглощения с 372 нм (УФ область). Возникает т. наз. разветвленная (или конкурирующая) сопряженная система (2 электронодонорных и 1 электроноакцепторный заместитель). Если же понизить электронодонорные свойства группы NH2 ацетилированием, конкурентоспособность ее уменьшается, длинноволновая полоса поглощения снова смещается батохромно почти до уровня гидрола Михлера (до
372 нм (УФ область). Возникает т. наз. разветвленная (или конкурирующая) сопряженная система (2 электронодонорных и 1 электроноакцепторный заместитель). Если же понизить электронодонорные свойства группы NH2 ацетилированием, конкурентоспособность ее уменьшается, длинноволновая полоса поглощения снова смещается батохромно почти до уровня гидрола Михлера (до 590 нм) и восстанавливается синий цвет.
590 нм) и восстанавливается синий цвет.
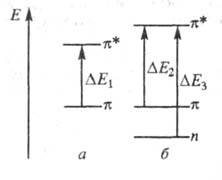
Рис. 2. Уровни энергии НМО и НСМО и электронные переходы в молекулах гидрола Михлера (а) и аурамина (б).
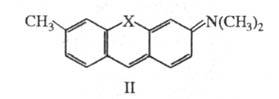
В случае, если 2 ближайших друг к другу атома С в бензольных кольцах гидрола Михлера замкнуть через электроотрицат. атомы N или О, образуется соед. формулы II. При этом энергия НМО не изменяется, а энергия НСМО повышается, что приводит к смещению длинноволновой полосы поглощения до 550 нм в случае X = О (пурпурный пиронин) и до 488 нм в случае X = NH (акридиновый оранжевый).
Большое влияние на поглощение света орг. соед. оказывают пространств. факторы, приводящие к искажениям формы молекул. При этом существенное значение имеет характер искажения. Если молекула перестает быть плоской, то происходит сдвиг в коротковолновую область, т. е. цвет повышается; если же происходит изменение валентных углов без существенного нарушения плоской формы молекулы, то имеет место углубление цвета. В первом случае причина повышения цвета связана с частичным или полным разобщением отдельных участков цепи сопряжения вследствие нарушения копланарности молекулы из-за поворота одних ее частей по отношению к другим вокруг простой связи. Например, молекулы дигидрофенантрена (III;
в коротковолновую область, т. е. цвет повышается; если же происходит изменение валентных углов без существенного нарушения плоской формы молекулы, то имеет место углубление цвета. В первом случае причина повышения цвета связана с частичным или полным разобщением отдельных участков цепи сопряжения вследствие нарушения копланарности молекулы из-за поворота одних ее частей по отношению к другим вокруг простой связи. Например, молекулы дигидрофенантрена (III;  267 нм) и перилена (IV;
267 нм) и перилена (IV; 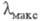 432 нм), имеющие плоскую форму, поглощают свет в более длинноволновой области, чем бифенил (V;
432 нм), имеющие плоскую форму, поглощают свет в более длинноволновой области, чем бифенил (V; 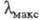 251,5 нм) и бинафтил (VI;
251,5 нм) и бинафтил (VI; 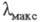 291 нм), у которых возможен поворот вокруг биарильной связи, нарушающий сопряжениеπ — электронов двух ароматич. ядер.
291 нм), у которых возможен поворот вокруг биарильной связи, нарушающий сопряжениеπ — электронов двух ароматич. ядер.
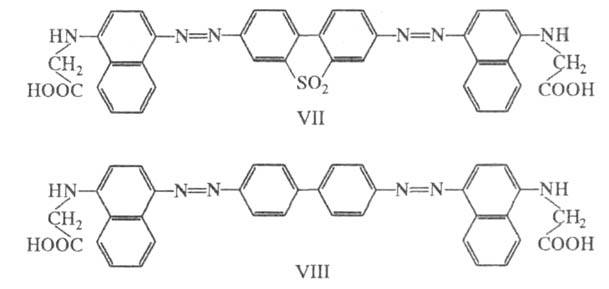
По той же причине из двух аналогичных азокрасителей производное бензидинсульфона (VII; синий) окрашено глубже, чем производное бензидина (VIII; коричнево-красный).
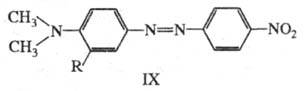
В азокрасителе IX введение заместителей в орто-положение к диалкиламиногруппе нарушает сопряжение неподеленной пары азота с -системой, что приводит к повышению цвета. Например, IX (R=H) поглощает при
-системой, что приводит к повышению цвета. Например, IX (R=H) поглощает при 475 нм; при R = CH3 или изо-C3H7
475 нм; при R = CH3 или изо-C3H7 438 и 420 нм соотв.; одновременно уменьшается интенсивность поглощения в 1,5 и 1,7 раза.
438 и 420 нм соотв.; одновременно уменьшается интенсивность поглощения в 1,5 и 1,7 раза.
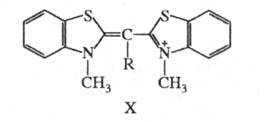
При искажении углов между направлениями связей атомов без значит. нарушения плоской структуры молекулы сопряжение -электронов существенно не нарушается, но возникающее напряжение сближает уровни энергии молекулы в основном и возбужденном состояниях, снижая тем самым энергию возбуждения. Так, введение в центральную метиновую группу монометинцианина (X; R = Н;
-электронов существенно не нарушается, но возникающее напряжение сближает уровни энергии молекулы в основном и возбужденном состояниях, снижая тем самым энергию возбуждения. Так, введение в центральную метиновую группу монометинцианина (X; R = Н;  425 нм) метильной группы (X; R = CH3;
425 нм) метильной группы (X; R = CH3;  465 нм), создающей пространственные затруднения, вызывает углубление цвета при одновременном падении интенсивности поглощения почти вдвое.
465 нм), создающей пространственные затруднения, вызывает углубление цвета при одновременном падении интенсивности поглощения почти вдвое.
Большое влияние на цвет орг. соед. оказывает присутствие в его структуре металла. При образовании комплекса создаются новые возможности электронных переходов, обусловливающие появление новых полос поглощения в спектрах комплексов. Появление этих полос связано с переносом электрона с высшей занятой МО (ВЗМО) орг. молекулы (лиганда) на своб. атомную орбиталь металла, с переходом ^-электрона металла на НВМО лиганда (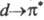 -переход), а также с возможностью
-переход), а также с возможностью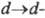 перехода, который возникает благодаря снятию вырождения с вакантных d-орбиталей металла под влиянием поля лиганда. Обычно
перехода, который возникает благодаря снятию вырождения с вакантных d-орбиталей металла под влиянием поля лиганда. Обычно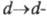 переходы существенно на цвет комплексов не влияют, т. к. их полосы большей частью находятся в ИК области спектра.
переходы существенно на цвет комплексов не влияют, т. к. их полосы большей частью находятся в ИК области спектра.
В химии красителей в качестве металлов-комплексообразователей наиб. часто используют Cr, Cu, Ni, Co, Fe, A1 в разл. степенях окисления, обычно 2 или 3. При образовании внутрикомплексных соед. атом металла входит в устойчивый 5- или 6-членный цикл; при этом он связывается с двумя разл. атомами, один из которых отдает ему неподеленную пару электронов (донорно-акцепторная, или координац., связь). Если эти электроны участвуют в системе сопряжения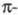 связей, ответственных за поглощение света, комплексообразование сопровождается углублением цвета, напр. цвет соед. XI изменяется от оранжевого до зеленого при образовании комплекса с Fe, до оливкового — с Cr, до красно-коричневого — с Со.
связей, ответственных за поглощение света, комплексообразование сопровождается углублением цвета, напр. цвет соед. XI изменяется от оранжевого до зеленого при образовании комплекса с Fe, до оливкового — с Cr, до красно-коричневого — с Со.
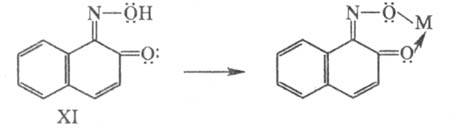
В случае, если неподеленные пары электронов не входят в систему сопряженных двойных связей, ответственных за возникновение окраски, комплексообразование увеличивает устойчивость окрасок к разл. воздействиям (света, тепла и др.).
Электронной теории цветности предшествовали более ранние теории. Одной из первых была хромофорно-ауксохромная теория О. Витта (1876), согласно которой окрашенные соед. содержат ненасыщ. группы — хромофоры (напр., N=N, NO2, NO, CH = CH, C = O), ответственные за цвет орг. соед. (такие соед. наз. хромогенами), и электронодонорные группы — ауксохромы (напр., OH, SH, NH2, NHR, NR2), повышающие интенсивность окраски. Несмотря на то, что теория Витта устарела, предложенная им терминология используется в совр. химии красителей.
Хиноидная теория цветности, созданная Г. Армстронгом и Р. Ниецким (1887), объясняла появление окраски перегруппировкой ароматич. (бензоидного) ядра в хиноидное.
Наиб. близка к совр. теории цветности теория Г. Льюиса (1916), по которой "цвет обусловлен селективным поглощением света валентными электронами, частоты которых синхронны с соответствующей частотой световых колебаний".
Лит.: Венкатараман К., Химия синтетических красителей, пер. с англ., т. 1, Л., 1956, т. 3, Л., 1974; Дьюар М., Догерти Р., Теория возмущений молекулярных орбиталей в органической химии, пер. с англ., М., 1977; Хедвиг П., Прикладная квантовая химия, пер. с англ., М., 1977; Барлтроп Дж., Коил Дж., Возбужденные состояния в органической химии, пер. с англ., М., 1978; Киприанов А. И., Цвет и строение цианиновых красителей, К., 1979; Степанов Б. И., Введение в химию и технологию органических красителей, 3 изд., М., 1984; Свердлова О. В., Электронныеспектры в органической химии, 2 изд., Л., 1985.
Б. И. Степанов
